The Challenge and Importance of Early Diagnosis
Jane Dion 2013
Reviewed by Dr. Christian Pagnoux
Churg-Strauss Syndrome is a challenging disease to diagnose and consequently, many patients are diagnosed in the later stages of the disease after permanent organ damage, and tragically, even death occur. This is such an important issue that one of the world’s most renown specialists in CSS/EGPA, Dr. Christian Pagnoux, has agreed to write about how to recognize and diagnose the disease in its earlier stages so that treating physicians such as pulmonologists, ER doctors, ENT physicians and others will perhaps consider the diagnosis before organ damage occurs. In addition, patients who are searching for clues about their “mystery illness” may find some answers about whether or not they should ask their doctors to consider CSS.
When CSS was first recognized as a disease entity back in the 1950s, diagnosis was made on autopsy and it was almost always fatal. Now, with more timely diagnoses and effective treatment, patients are living long healthy lives, even though most remain on some type of medication and relapses are common. Early diagnosis is the key and its importance cannot be overstated.
Sometimes, despite an early diagnosis, one can not (always) prevent some of the most severe disease manifestations such as cardiomyopathy or neuropathy, because these are more often manifestations of a severe disease rather than progression of a disease whose diagnosis was too delayed. Some patients start their disease with sudden cardiac arrest after years of a common usual asthma.
The CSS message boards are filled with patients living with painful neuropathy, heart conditions, kidney damage, and lung damage which could have been, for some of them, more limited with an earlier diagnosis. Again, this underscores the importance of finding ways to diagnose CSS/EGPA early.
Following is the article written by Dr. Pagnoux that explains the disease process and gives extremely helpful guidelines to aid in diagnosing this complex and complicated disease.
EGPA: early diagnosis is better
By Christian Pagnoux, MD MPH MSc
May 2013 Mount Sinai Hospital Division of Rheumatology – Vasculitis clinic
60 Murray Street Room 2-220 Toronto, ON, M5T 3L9, Canada
Phone: 416-586-4800 ext. 8549
Fax: 416-586-8766
[email protected]
http://www.canvasc.ca
Article in PDF format
As compared with initial descriptions of eosinophilic granulomatosis and polyangiitis (EGPA) showing poor survival, patient outcomes have dramatically improved over the past 2 decades. At 1 and 5 years post-diagnosis, survival rates now exceed 90% and 85%, respectively [1-3]. Besides advances in the therapeutic management of EGPA, better awareness and recognition of this rare condition have clearly contributed to improved survival. Delayed diagnosis and initiation of appropriate treatments can indeed negatively affect overall prognosis and outcomes.
However, it should be acknowledged that the diagnosis of EGPA often remains challenging and in some cases can be confirmed only during the course of the disease. EGPA is a rare disease with an incidence of about 1 to 2 per million people and a prevalence of about 10 to 15 per million [4-6]. Hence, most physicians will not likely see more than a couple of EGPA cases during their career.
Diagnostic criteria to help with early diagnosis are lacking. The 1990 American College of Rheumatology criteria included the most typical “full-blown” disease characteristics and aimed at classifying patients with already diagnosed and proven vasculitis into a disease category. The Chapel Hill nomenclature are useful to define EGPA among the medical community but cannot be used for diagnosis. More importantly, we lack a good biologic or radiologic test to detect early EGPA or confirm the diagnosis. Anti-neutrophil cytoplasm antibodies (ANCA) are present in only 30% to 40% of EGPA patients at diagnosis and/or during disease flare (mainly antimyeloperoxidase perinuclear ANCA) [1, 7-9]. Increased blood eosinophil count can suggest disease but is lacking in specificity in that it can be observed in other conditions as well, including simple allergy. Serum level of immunoglobulin E (IgE) is also usually elevated, but this sign lacks specificity even more so than blood eosinophilia. Biopsy of an affected tissue thus remains the gold standard to support the diagnosis; it may show vasculitis, i.e. inflammation of the blood vessel wall, mainly with eosinophils, with other more subtle but inconstant features such as fibrinoid necrosis or granulomas. However, biopsies can be invasive and, depending on the organ, can cause moderate but irreversible damage such as persistent localized numbness after a peripheral nerve biopsy. In addition, biopsies do not always show typical histologic features, depending on the organ and because vasculitis lesions are segmental (i.e., have a patchy distribution along blood vessels).
Hence, physicians should think earlier than later of the possibility of EGPA in several clinical settings described below and must not hesitate to refer patients to a vasculitis center for evaluation and further investigation if needed. As described by Lanham et al., in the early 1980s, EGPA may be divided into and progress in 3 stages [10]: asthma and/or recurrent nasal polyposis (stage 1), which indeed corresponds more to a background condition than EGPA if not a prerequisite to 3 development of blood and tissue eosinophilia (stage 2), then EGPA with its vasculitis manifestations (stage 3). Early identification of these pre-EGPA stages may help prevent severe complications of EGPA and promote faster remission. However, not all patients will show progression through these 3 stages, and one should not over- or prematurely diagnose EGPA. Eosinophilic allergic asthma with nasal polyposis is indeed more frequent than EGPA, and EGPA will not develop in most patients with allergic asthma [11].
The most frequent clinical settings that should alert physicians (and patients) to the possibility of EGPA are relatively easy to identify and remember.
- Asthma, especially late-onset asthma (i.e., starting in adulthood), that gradually worsens and becomes refractory to usual antiasthma drugs, with increased eosinophilia on white blood cell count. In most of these cases, allergic asthma or allergic bronchopulmonary aspergillosis is diagnosed, but early stages of EGPA can present in this way.
- Recurrent bronchitides and/or “pneumoniae” in a patient with background asthma, especially late-onset asthma, with increased eosinophilia on white blood cell count. In most of these cases, “simple” infection, allergic bronchopulmonary aspergillosis or eosinophilic pneumonia is diagnosed, but early stages of EGPA can present in this way.
- Worsening, lingering and/or recurrent sino-nasal polyposis and/or sinusitis, especially if associated with (late-onset) asthma, with increased eosinophilia on white blood cell count. These manifestations are not sufficient for a diagnosis of EGPA because of no vasculitis, but early stages of EGPA can present in this way.
- Recurrent skin rash (any type) or hives with increased eosinophilia on white blood cell count. In most of these cases, simple allergy or chronic urticaria is diagnosed, but early 4 stages of EGPA can present in this way and many different types of skin lesions can occur in EGPA (Figures 1 to 4).
- In a patient with asthma and/or sino-nasal polyposis, any symptoms of systemic vasculitis, including skin purpuric rash, numbness, tingling or weakness in hands or feet (mononeuritis multiplex), scleritis (or episcleritis), or renal disease (microscopic hematuria being the first manifestation of glomerulonephritis). Other possible and/or more severe manifestations, such as coronary arteritis or gut perforations due to inflammation and occlusion of the small vessels of the bowels, are rare features of EGPA that are more easily considered related to vasculitis (EGPA or another type of vasculitis).
- In a patient with asthma and/or sino-nasal polyposis, any new or worsening general or constitutional symptoms, including fever, joint pain, diffuse muscle pain, major involuntary weight loss, chest pain, palpitations or abdominal pain. These symptoms are not specific but may be the first signs of a vasculitis, including EGPA.
In such cases, it is wise to control blood cell count, including eosinophil count; check some inflammatory signs such as level of C-reactive protein (CRP) and/or erythrocyte sedimentation rate (ESR); and perhaps order an ANCA screening test as well as other investigations according to the clinical presentation (e.g., chest X-ray or CT with respiratory symptoms; CT scan of sinuses with ear nose, and throat manifestations [Figure 5]; electromyography of peripheral nerve conduction with numbness; electrocardiography and cardiac imaging). Biopsies of skin lesions are easy to perform but may show non-specific leucocytoclastic vasculitis. The sensitivity of sinus biopsy is low (<50%). Other biopsies, such as peripheral nerve or lung lesion, may be considered depending on the clinical presentation and after review of all obtained results, which may be sufficient to consider the diagnosis of EGPA highly probable for starting the appropriate treatment.
As with other vasculitides, such as giant cell arteritis, starting systemic corticosteroid treatment early, which remains the cornerstone of EGPA treatment, should not be considered a mistake, at least after all the appropriate diagnostic investigations have been realized (especially to rule out parasitic infections that can cause blood eosinophilia). Corticosteroids alone may still be insufficient to prevent more severe vasculitis complications, such as those listed in the original 1996 five factor score (i.e., cardiomyopathy or central nervous system, severe gastrointestinal or renal involvement [creatinine level > 140 µmol/l or proteinuria > 1 g/24 hr) [12]. Physicians should remain alert to such severe complications and follow patients closely for early detection. Conversely, corticosteroid treatment should not be prolonged in the absence of a clear diagnosis. The diagnosis of EGPA should not be made too easily (and erroneously) for the (rare) patients with allergic asthma who depend on systemic corticosteroids for asthma control but who never had any other (vasculitis) manifestations, simply because they are corticosteroid-dependent. A corticosteroid-sparing agent can still be considered for such patients with intractable asthma. Of note, the discontinuation of corticosteroids in these patients with intractable asthma, sometimes after a successful trial with other anti-asthma drugs such as a leukotriene receptor antagonist, may unmask vasculitis manifestations (i.e., a “forme fruste” of EGPA) [13-15].
Studies may eventually identify more specific and sensitive biologic markers that will help in the early diagnosis of EGPA and in differentiating EGPA from mimicking diseases such as common allergic asthma, allergic bronchopulmonary aspergillosis, eosinophilic pneumonia or primary hypereosinophilic syndrome. Several potential biologic candidates have been investigated and 6 include eotaxin-3 (CCL26), interleukin 5 (IL-5), IL-25, eosinophil cationic protein, and thymus and activation-regulated chemokine (TARC or CCL17) but with relatively disappointing results or that need to be validated in a larger number of patients with EGPA or mimicking conditions [16-19]. Because EGPA is a rare disease, achieving reliable and reproducible results will take some time. Until then, effort must continue to improve awareness of this condition, systematize the diagnostic approach and investigations, optimize treatments for severe manifestations, reduce cumulative corticosteroid exposure and limit treatment-related side effects.
Figures 1-5
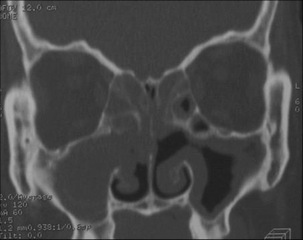


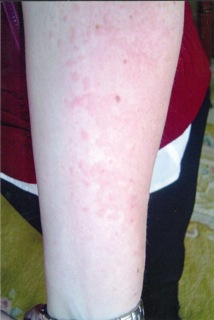
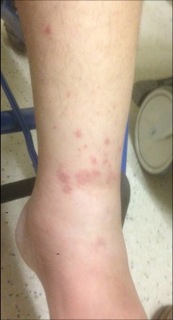
Figure 1: CT scan of sinuses showing bilateral maxillary and sphenoidal sinusitis and nasal polyps in a patient with EGPA diagnosed after she presented some vasculitis manifestations (lung infiltrates and nodules with eosinophilic vasculitis on lung biopsy).
Figures 2 to 5: Possible lesion types in EGPA: purpuric and ulcero-necrotic lesions on both legs (2), diffuse erysipelas-like rash with subcutaneous nodules (circle) on one leg (3), pseudo-urticarial hive-like (itchy) lesions on one arm (4), and macular erythematous and purpuric rash on one leg (5) from 4 different patients.
References
1. Comarmond, C., et al., Eosinophilic granulomatosis with polyangiitis (Churg-Strauss):
clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis
Study Group cohort. Arthritis Rheum, 2013. 65(1): p. 270-81.
2. Pagnoux, C., Churg-Strauss syndrome: evolving concepts. Discov Med, 2010. 9(46): p. 243-52.
3. Moosig, F., et al., A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): monocentric experiences in 150 patients. Ann Rheum Dis, 2013. 72(6): p. 1011-7.
4. Mohammad, A.J., et al., Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford), 2007. 46(8): p. 1329-37.
5. Pagnoux, C. and L. Guillevin, Churg-Strauss syndrome: evidence for disease subtypes? Curr Opin Rheumatol, 2010. 22(1): p. 21-8.
6. Vaglio, A., C. Buzio, and J. Zwerina, Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy, 2013. 68(3): p. 261-73.
7. Sinico, R.A., et al., Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum, 2005. 52(9): p. 2926-35.
8. Keogh, K.A. and U. Specks, Churg-Strauss syndrome: clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med, 2003. 115(4): p. 284-90.
9. Baldini, C., et al., [Churg-Strauss syndrome: outcome and long-term follow-up of 38 patients from a single Italian centre]. Reumatismo, 2009. 61(2): p. 118-24.
10. Lanham, J.G., et al., Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore), 1984. 63(2): p. 65-81.
11. Harrold, L.R., et al., Incidence of Churg-Strauss syndrome in asthma drug users: a population-based perspective. J Rheumatol, 2005. 32(6): p. 1076-80.
12. Guillevin, L., et al., Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore), 1996. 75(1): p. 17-28.
13. Wechsler, M. and J.M. Drazen, Churg-Strauss syndrome. Lancet, 1999. 353(9168): p. 1970-1.
14. Wechsler, M.E., et al., Churg-Strauss syndrome in patients receiving montelukast as treatment for asthma. Chest, 2000. 117(3): p. 708-13.
15. Wechsler, M.E., et al., Churg-strauss syndrome in patients treated with omalizumab. Chest, 2009. 136(2): p. 507-18.
16. Khoury, P., et al., Serum biomarkers are similar in Churg-Strauss syndrome and hypereosinophilic syndrome. Allergy, 2012. 67(9): p. 1149-56.
17. Polzer, K., et al., Eotaxin-3 is involved in Churg-Strauss syndrome–a serum marker closely correlating with disease activity. Rheumatology (Oxford), 2008. 47(6): p. 804-8.
18. Zwerina, J., et al., Eotaxin-3 in Churg-Strauss syndrome: a clinical and immunogenetic study. Rheumatology (Oxford), 2011. 50(10): p. 1823-7.
19. Hellmich, B., E. Csernok, and W.L. Gross, Proinflammatory cytokines and autoimmunity in Churg-Strauss syndrome. Ann N Y Acad Sci, 2005. 1051: p. 121-31.
Figure legends
Figures 1 to 4: Possible lesion types in EGPA: purpuric and ulcero-necrotic lesions on both legs (1), diffuse erysipelas-like rash with subcutaneous nodules (circle) on one leg (2), pseudourticarial hive-like (itchy) lesions on one arm (3), and macular erythematous and purpuric rash on one leg (4) from 4 different patients.
Figures 5: CT scan of sinuses showing bilateral maxillary and sphenoidal sinusitis and nasal polyps in a patient with EGPA diagnosed after she presented some vasculitis manifestations (lung infiltrates and nodules with eosinophilic vasculitis on lung biopsy).
In Loving Memory of Those Lost to CSS/EGPA
The CSSA occasionally receives heartbreaking stories from those who have lost loved ones to the disease. Some families and friends fundraise for the CSSA in their loved ones honor. Following are just a few of the real people lost to this disease. In addition, Tina Bennett, who lost her 21 year old daughter Erika to CSS, writes about the pain of seeing her child succumb to an illness that the hospital and doctors did not know she had until after an autopsy was performed. Tina honors her daughters memory by holding fundraisers in her honor. The CSSA is very grateful for her efforts and her belief in our mission.
Dr. Carole Disendof
 September 6, 1951 – October 1, 2011
September 6, 1951 – October 1, 2011
She was a Beverly Hills psychologist for over 25 years and the best selling author of “Talk the Weight Off! She was the most caring, supportive, generous and loving person to her family, friends and clients. According to the obituary, Carole was very instrumental in changing many lives.
Frank Ranelli, Jr.
 Frank died at age 69. He proudly served his country in the U.S. Army. Frank was employed as a carpenter for over 30 years. He loved to read, fish and make his own fishing lures. He enjoyed attending all of his grandchildren’s activities.
Frank died at age 69. He proudly served his country in the U.S. Army. Frank was employed as a carpenter for over 30 years. He loved to read, fish and make his own fishing lures. He enjoyed attending all of his grandchildren’s activities.
Ralph E. Fago
Ralph E. Fago, of New York, died at age 30. Ralph was an Aerospace Systems Engineer with Lockheed Martin in Owego. He was also an accomplished musician, performing in venues throughout New York State. Ralph also spent his time as a volunteer firefighter. Ralph had many, many friends who miss him still and have held fundraisers in his memory.
Erika Machelle Bennett
 Lovey, Sissy, Hank, Baby
Lovey, Sissy, Hank, Baby
March 23, 1989 to August 14, 2010
By Tina Bennet
This is Erika’s story of Churg Strauss Syndrome. This is intended to be helpful information. Sadly, Erika’s diagnosis of CSS did not occur until after her passing, August 14 2010. At the initial time of passing the medical team had stated that it was Asthma and Pneumonia. *note: the final autopsy report indicated cause of death was from diffuse involvement of Churg-Strauss Syndrome in the heart, the lungs and the kidneys. (JD)
Erika was a very healthy, athletic young lady. In high school Erika excelled in basketball, volleyball and was an Honor student. Erika had a passion for music and art, she was active in our local church and she ate extremely healthy. Erika graduated from High school in 2007.
In the fall of 2007, Erika attended West Virginia University in hopes of an education in the medical field. In the fall of 2007, Erika began having breathing problems and sinus, allergy symptoms. Erika had seen local Asthma specialist and was tested for allergies.
During 2008 and 2009 her symptoms kept getting worse even though she continued school and worked as a waitress. In the fall of 2009 she was having such a hard time and feeling so bad she came home to Paden City WV to live with me, her mom, and attend a local community college. In the fall of 2009, Erika had tubes put into her ears to try to help with all the ear pain she was experiencing. Erika said her hearing felt like she was chewing up potato chips with your hands cupped over your ears. I tried that, and I could not believe that is what her hearing felt like.
In the spring of 2010, Erika had another set of tubes put into her ears this was in early spring just before her twenty first birthdays. In July, Erika found out she was expecting a child. I was so concerned about her health but I tried to be supportive and positive about the unexpected news. In July she went to a local obstetrician the baby’s heartbeat was fine, but I did tell the Doctor about my concerns over Erika’s health and how bad her asthma was.
In early August, Erika went to a local hospital having hard time breathing, covered in a red raised rash and ear pain. The emergency room Doctor put his hand on her shoulders turned her around, told her to look at the “Emergency Room” sign and said, “This is a place for sick people, people with real problems not rashes.”On August ninth 2010, Erika went to the Emergency room in Morgantown WV. She was admitted with a very low blood pressure. Erika still had the rash on her and was having extreme breathing problems. I left work at a Radio station in Parkersburg WV and went straight to the hospital. After the Doctor came in and talked to her, I took the Doctor aside and explained that there was a very large history of Rheumatoid Arthritis in my family. I said that my mother had passed at age 54 due to the complications of a lifetime of the disease. Both of my maternal grandmothers passed away due to this disease one only 34 and the other at age 61.
On August 10, the special disease Doctor said that there were no signs of RA. Throughout the week they continued to treat Erika for Pneumonia and gave her breathing treatments. The treatments became more frequent and not as affective. Erika’s blood pressure kept going down. By the evening of August 13th, they moved Erika to the Intensive care unit. The Doctor decided to transport Erika to the other Hospital in Morgantown WV that was more specialized in issues with the heart.
The last thing I said to Erika was that I will see you at the next hospital as I held her hand. I prayed endlessly all week for Erika. Staff of the hospital would ask to pray with us and I gladly accepted. I could tell Erika was worried about the baby but I was worried about my baby. I still believed Erika was going to pull through this. But as I rubbed her legs and felt how cold they were and saw her gasp and struggle for every breath. It felt like I was standing there watching her drown and I could not jump in and save her. I begged God, let me trade places with her, let it be me. There is no great pain than to see your child die right before your eyes. Earlier the Doctor had handed me a rosary, I clutched it as tight as we watched through the glass panels as a team of Doctors worked and worked on her to keep her here. Her family, watching through the glass praying, yelling for her to hang on, stay here. To this day I whisper to her, “Please don’t leave me, Lovey.”
 Early on the morning of August 14th 2010, I heard the Doctor Call time on my only Daughter, my Lovey, Erika Machelle Bennett. I ran from the room to the waiting room feel to my knees. It felt like somebody stabbed me with a knife, I couldn’t breathe, I tried to hurt myself and I knew there was no way I could live without my best friend my beautiful wonderful Daughter. The staff and Doctor held me for what seemed like forever. I had been given some medications, I was in shock. How can a person still be alive when it feels like someone took a shotgun and blew their heart apart?
Early on the morning of August 14th 2010, I heard the Doctor Call time on my only Daughter, my Lovey, Erika Machelle Bennett. I ran from the room to the waiting room feel to my knees. It felt like somebody stabbed me with a knife, I couldn’t breathe, I tried to hurt myself and I knew there was no way I could live without my best friend my beautiful wonderful Daughter. The staff and Doctor held me for what seemed like forever. I had been given some medications, I was in shock. How can a person still be alive when it feels like someone took a shotgun and blew their heart apart?
We, as a family, Erika’s two brothers Gabriel and Derrick and all the others went into the room where everyone had been working on her. She lay there, cold peaceful my precious baby. I kissed her I told her how much I love her and how proud I am of her and I said goodbye. Good by Lovey, we will meet again in Heaven. I love you Erika!